LIMMA Diagnostic Plots
Tyler Sagendorf
01 May, 2024
Source:vignettes/articles/limma_diagnostics.Rmd
limma_diagnostics.RmdThis article generates multidimensional scaling (MDS) and mean–variance trend plots (Extended Data Fig. 8A–G).
# Required packages
library(MotrpacRatTraining6moWATData)
library(MotrpacRatTraining6moWAT) # theme_pub, ggplotMDS
library(Biobase)
library(edgeR)
library(dplyr)
library(ggplot2)
save_plots <- dir.exists(paths = file.path("..", "..", "plots"))
# Transcriptomics pre-processing
# Convert filtered counts to normalized log2 counts-per-million reads
dge <- DGEList(counts = exprs(TRNSCRPT_EXP),
samples = pData(TRNSCRPT_EXP),
group = TRNSCRPT_EXP$exp_group)
dge <- calcNormFactors(dge, method = "TMM")
m_dge <- TRNSCRPT_EXP
exprs(m_dge) <- cpm(dge, log = TRUE)Multi-dimensional Scaling Plots
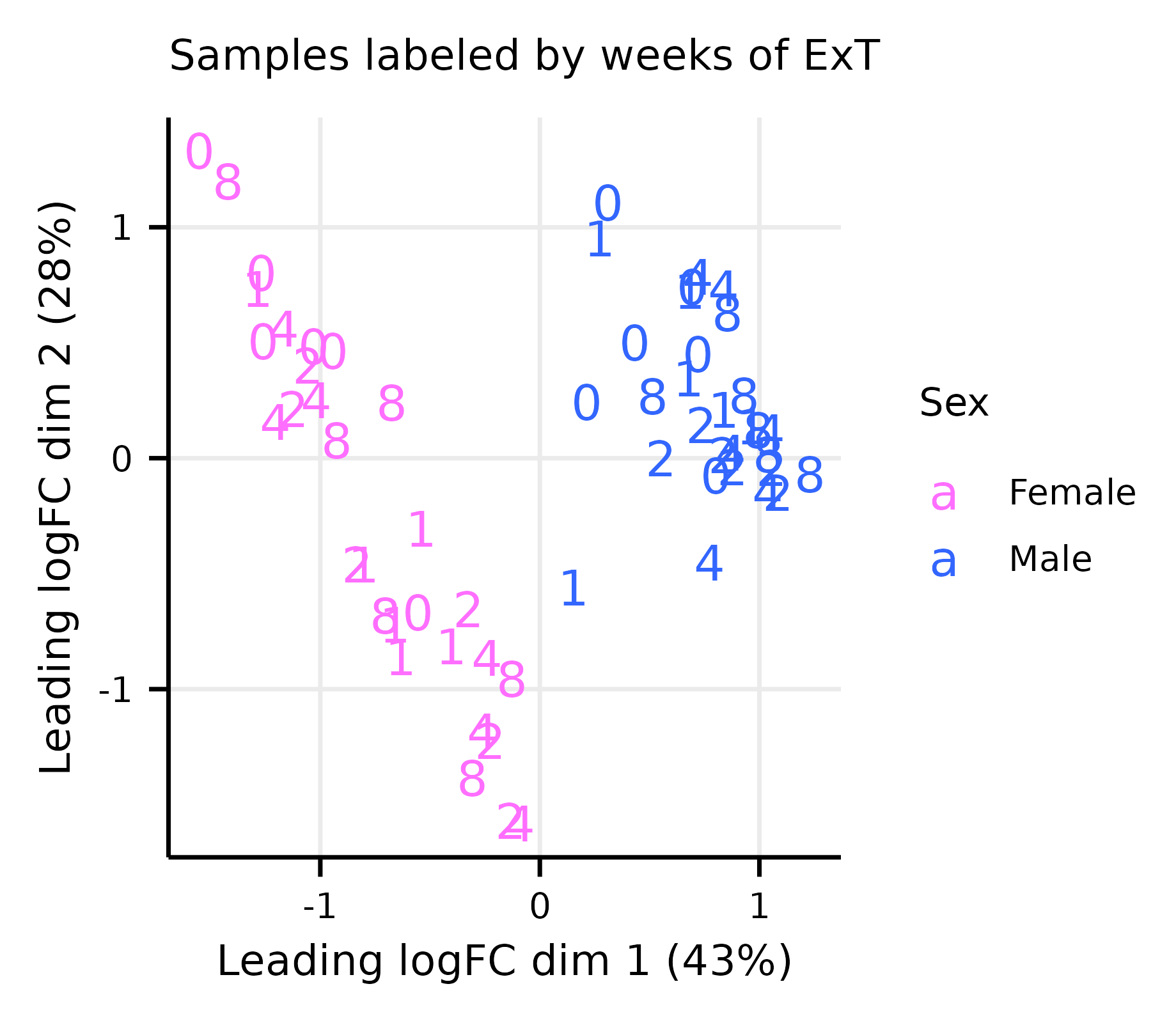
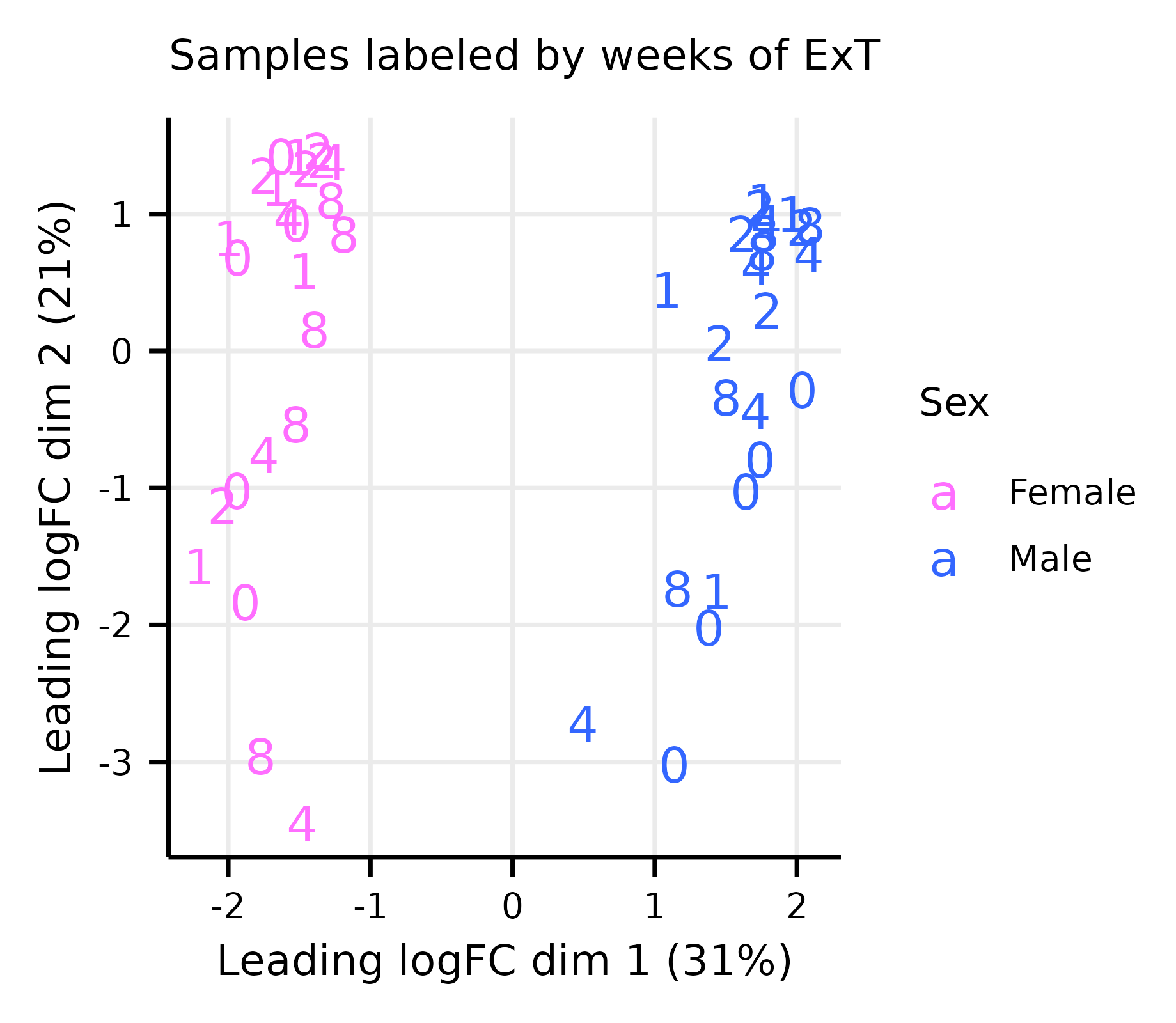
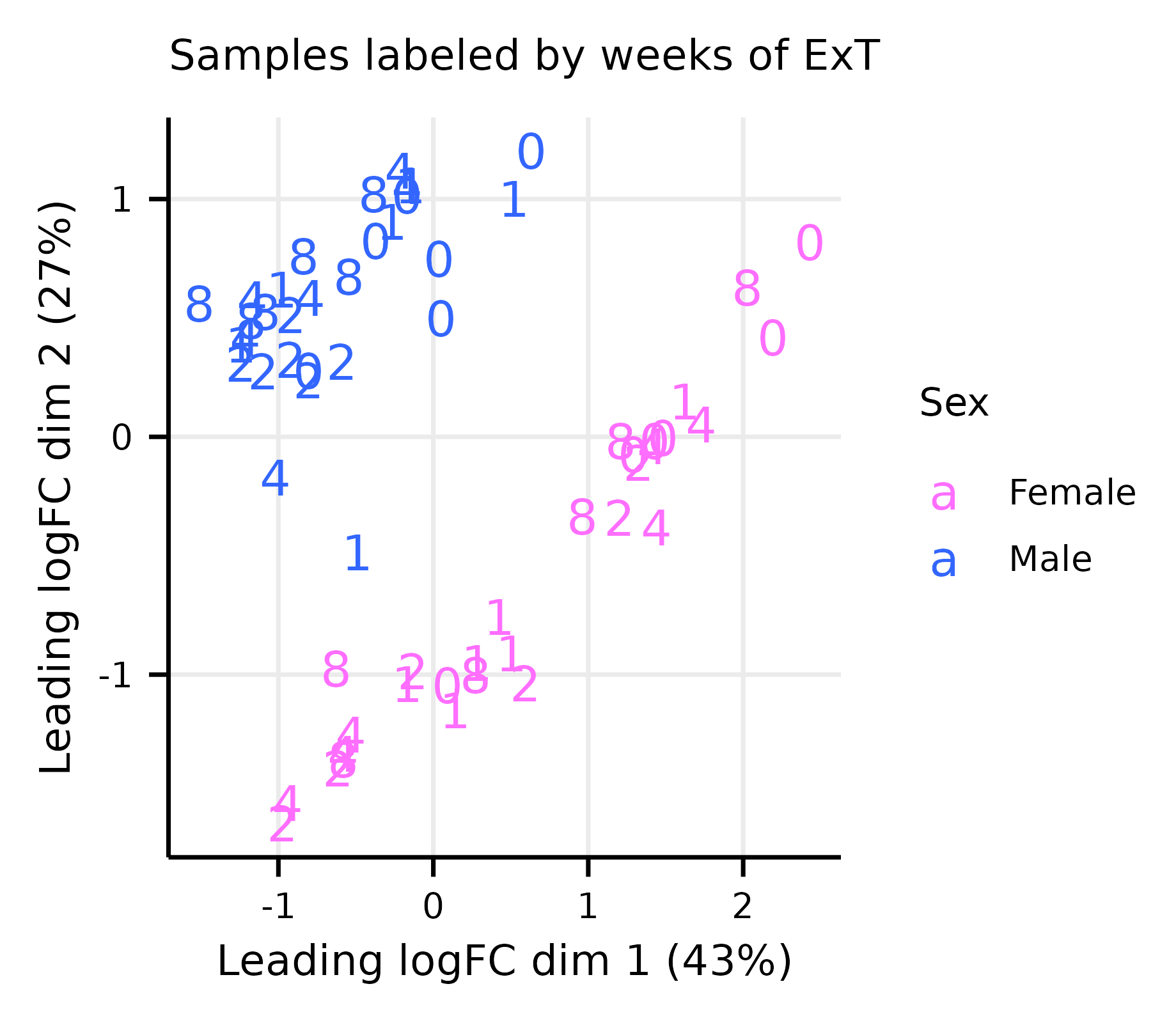
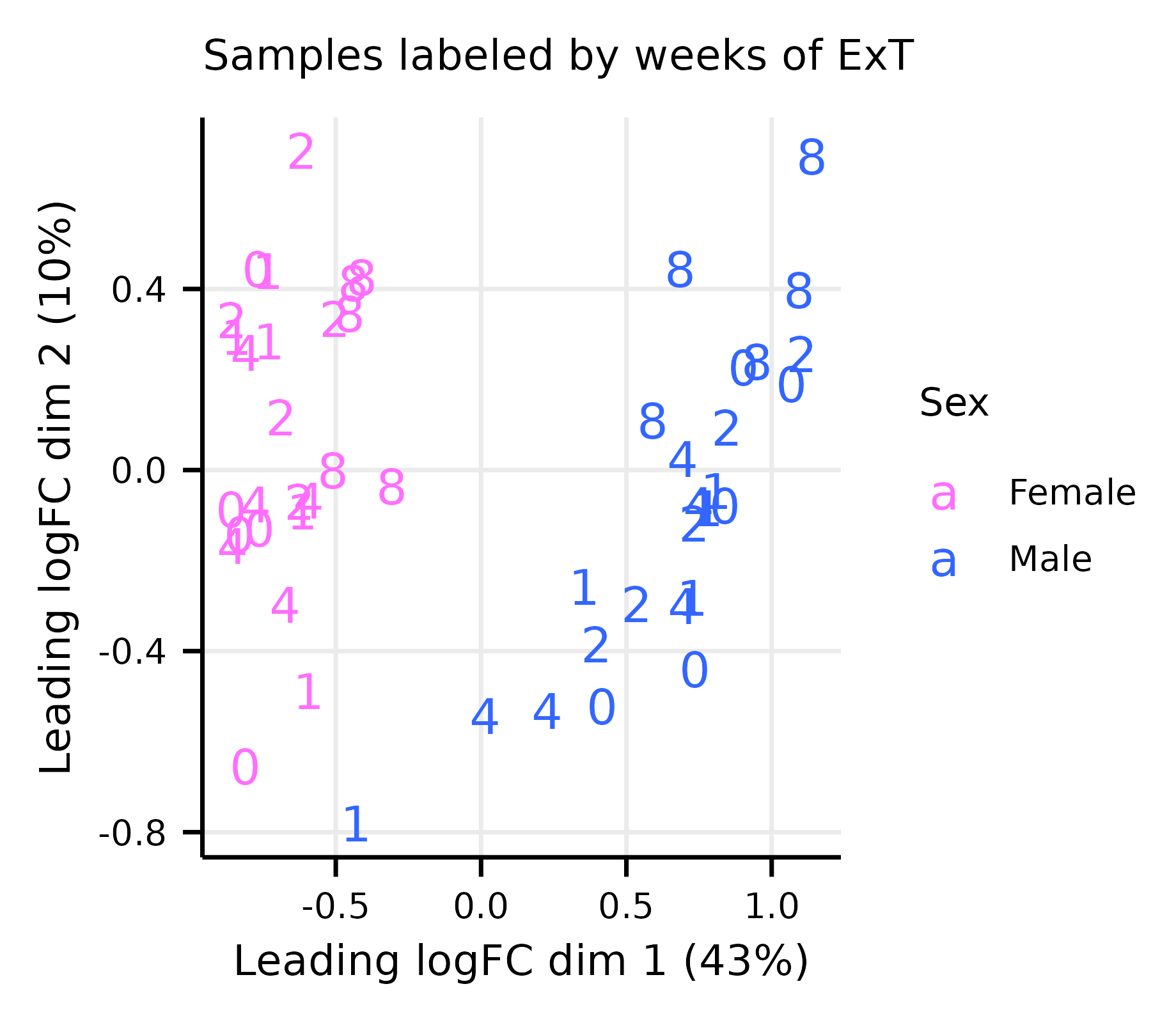
Mean–variance trend
# Do not run the following chunks when knitting
knitr::opts_chunk$set(eval = FALSE)PROT and PHOSPHO
# PROT and PHOSPHO:
# In vignettes/WATSC_DA.Rmd,
# 1) debug limma_full. Set
# plot = TRUE. Run all code up through the creation of fit.smooth.
# 2) debug limma::plotSA. Run plotSA(fit.smooth) up until the plot is made.
# 3) Set working directory to the location of this article and run the following
# code. Modify the -ome in the file name as needed (either PROT or PHOSPHO).
ome <- "PROT" # PROT or PHOSPHO
# Create data.frame so that red points (outliers) will be plotted on top
mv_prot_df <- data.frame(x = x,
y = y,
color = colv,
s = sqrt(sqrt(fit$s2.prior))) %>%
mutate(color = factor(color, levels = c("black", "red"),
labels = c("Normal", "Outlier"))) %>%
arrange(color)
# Plot
mv_prot <- ggplot(mv_prot_df) +
geom_point(aes(x = x, y = y, color = color),
size = 1, alpha = 0.5, shape = 16) +
geom_line(aes(x = x, y = s), linewidth = 0.3,
color = "red", lty = "dashed") +
coord_cartesian(ylim = c(0, 3)) +
scale_y_continuous(expand = expansion(mult = c(5e-3)),
limits = c(0, NA)) +
scale_color_manual(name = NULL,
values = c("black", "red"),
labels = c("Normal", "Outlier")) +
labs(x = "Average log-expression",
y = expression(sqrt("sigma")),
subtitle = "eBayes(trend=TRUE, robust=TRUE)") +
theme_pub() +
theme(axis.line.y.right = element_blank(),
strip.text = element_text(hjust = 0,
margin = margin(t=0, r=0, b=5, l=0)),
legend.key.size = unit(6, "pt"),
# legend.margin = margin(r = 0, l = 0),
legend.position = c(0.8, 5/6),
legend.background = element_rect(fill = "white", color = "black",
linewidth = 0.3),
panel.grid.minor = element_line(linewidth = 0))
ggsave(
filename = sprintf(file.path("..", "..", "plots", "limma_diagnostics_mean-variance_%s.pdf"), ome),
mv_prot, height = 2, width = 2.3, dpi = 400, bg = "white"
)
# exit debugger nowTRNSCRPT
# In vignettes/WATSC_DA.R:
# 1) debug `limma_full`. Run all code before `limma::voomWithQualityWeights`
# is called.
# 2) debug `limma::voomWithQualityWeights`. Set plot=TRUE. Run all code before
# the second use of `voom`.
# 3) debug `voom`. Run all code before `plot(sx, sy, ...)`
# 4) Run the following code:
mv_trnscrpt <- ggplot(mapping = aes(x = sx, y = sy)) +
geom_point(alpha = 0.3, size = 1, shape = 16) +
geom_smooth(formula = y ~ x, method = "loess",
linewidth = 0.3,
color = "red", lty = "dashed") +
labs(x = expression(paste("log"[2],"(count size + 0.5)")),
y = expression(sqrt("standard deviation")),
subtitle = "voomWithQualityWeights(..., plot=TRUE)") +
scale_y_continuous(limits = c(0, 2.5),
expand = expansion(mult = 5e-3)) +
theme_pub() +
theme(axis.line.y.right = element_blank(),
strip.text = element_text(hjust = 0,
margin = margin(t=0, r=0, b=5, l=0)),
legend.key.size = unit(6, "pt"),
legend.margin = margin(r = 0, l = 0),
panel.grid.minor = element_line(linewidth = 0))
ggsave(file.path("..", "..", "plots", "limma_diagnostics_mean-variance_TRNSCRPT.pdf"),
mv_trnscrpt, height = 2, width = 2.3, dpi = 400, bg = "white")
# exit debugger now